Jack
A. Cook, Ph.D. and Howard N. Bockbrader, Ph.D.
Clinical Pharmacokinetics and Pharmacodynamics Department,
Pfizer Global Research and Development, Ann Arbor, MI
Introduction
In 1995 Amidon et al. devised a biopharmacetics classification
system (BCS) to classify drugs based on their aqueous solubility
and intestinal permeability(1). It was then recognized that dissolution
rate has a negligible impact on bioavailability of highly soluble
and highly permeable (BCS Class I) drugs when their formulation's
dissolution is sufficiently rapid(2). As a result, various regulatory
agencies including the United States Food and Drug Administration
(FDA) now allow bioequivalence of formulations of BCS Class I
drugs to be demonstrated by in vitro dissolution (often called
a biowaiver)(3,4). This paper examines one approach to the integration
of the biopharmaceutics classification system in drug development.
Potential Cost
Savings
Before attempting to implement any new paradigm, it is prudent
to examine the potential savings of the paradigm relative to the
costs of implementation. The potential savings is a function of
the potential number and likely cost of bioequivalence studies
saved (not performed). The cost of implementation is a function
of the number and the costs of compound and formulation characterizations.
To examine the potential savings,
the number of bioequivalence studies performed by the pharmaceutical
industry per year was examined. Between January 1998 and May 2001,
there were 229 New Drug Applications (NDAs) and 466 Abbreviated
New Drug Applications (ANDAs) approved by the FDA for oral immediate
release products (5). Recently, DiMasi noted an approval rate
of approximately 90% for NDAs (6). Assuming similar approval rates
for ANDAs, annual submission rates over the same period were 74
NDAs/year and 152 ANDAs/year. The Product Quality Research Institute
has noted that the average NDA submission contains 3 to 6 in vivo
bioequivalence studies (7). If one assumes that ANDAs contain
1 in vivo bioequivalence study, this suggests that industry performs
approximately 350 to 600 bioequivalence studies each year. The
authors have estimated that a typical bioequivalence study costs
approximately $250,000 including costs for packaging, clinical
facilities, data internalization, bioanalytical analysis, pharmacokinetic
and statistical analysis, and report generation. Thus, it is estimated
that the pharmaceutical industry spends between 90 and 150 million
dollars a year on bioequivalence studies. Further, this must be
recognized as an underestimate as the data does not capture bioequivalence
studies performed on drugs for which NDAs or ANDAs are never filed.
To determine the fraction of drugs likely amenable to biowaivers, NDAs for oral immediate release prescription products approved between January 1998 and May 2001 were characterized based on literature data and the product label. Each compound was counted only once when multiple formulations were approved. A compound was considered highly permeable if data existed that suggest it meets permeability criteria listed in the FDA BCS guidance (3). Data on solubility was generally limited and precluded precise classification. Therefore, a compound was classified as highly soluble if it met guidance criteria (highest dosage strength soluble in 250 mL) at one pH between 1 and 7.5 and was not precluded (i.e. highest dosage strength not soluble) from being highly soluble at some other pH.
Using the above criteria, approximately
25% of all compounds were classified as highly soluble and permeable
with approximately another 41% having insufficient data to allow
classification.
Using the 25% estimated above, there is the potential to save
one quarter the annual expenditures on bioequivalence studies,
$22 to $38 million dollars/year. Additional indirect savings can
occur if bioequivalence studies are rate limiting to drug development.
For example, suppose that results of a bioequivalence study are
needed before proceeding with development of a compound with eventual
peak sales of one billion dollars/year. It is not unreasonable
to assume that results of in vitro dissolution can be obtained
6 weeks earlier than results from an in vivo bioequivalence trial.
This time savings translates into a potential additional $110
million dollars in sales from a 6 week earlier approval. Further,
by not having to run a human bioequivalence trial, clinical resources
are freed to be applied elsewhere.
The authors have estimated that
BCS characterization costs approximately $10,000 to $60,000 per
compound, depending on the method used to characterize permeability.
Additionally, it is estimated that it costs approximately $15,000
to characterize the dissolution of each formulation. Given that
the majority of compounds are not biowaiver candidates, a strategy
of formal classification of every compound in development would
quickly consume and may surpass any potential savings. Thus a
more targeted strategy is proposed during development to try to
optimize the potential savings.
An Implementation
Strategy
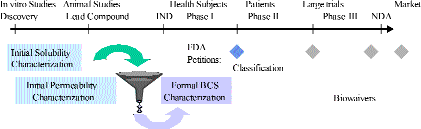
A targeted implementation strategy is illustrated in Figure
1. High throughput screens are used early in the drug discovery
process, when series of compounds are being synthesized to elucidate
structure activity relationships between compounds and the cell
target (8). An initial classification for permeability is gathered
using an automated permeability determination using CaCo2 on well
plates. A high throughput quasi-equilibrium method solubility
is performed at pH 6.5 using UV-detection. With these assays over
100 compounds can be characterized in a week. Compounds are considered
likely highly soluble if the projected human dose is soluble in
250 mL of water at pH 6.5 and likely highly permeable if the CaCo2
determined permeability is greater than that of metoprolol, which
was chosen as the standard by virtue of it being the high permeability
drug with the lowest permeability. It should also be noted that
a compound could also be considered likely highly permeable if
subsequent preclinical pharmacokinetic studies result in an absolute
bioavailability of 90% or more or if oral mass balance studies
result in 90% or more of the total radioactive dose recovered
in the urine.
Figure 1. An Industrial Implementation Strategy for the Biopharmaceutics Classification System.
Definitive BCS classification is
done when a potential Class I candidate enters human testing.
Classification is done according to methods outlined in the FDA
guidance (3). For instance, solubility is determined at pH 1.2,
7.5 and at pHs around the compounds pKa (pKa-1, pKa, and pKa+1).
In order of preference, permeability classification is determined
from renal excretion data from human studies, extent of renal
radioactivity excretion from human mass balance studies, human
absolute bioavailability, and preclinical permeability studies
(rat intestinal perfusion or CaCo2). The order of preference was
derived by first noting that human data is considered more definitive
than preclinical or in vitro data and second by the cost to obtain
the data. If the compound meets all criteria, a petition is sent
to the FDA asking for their agreement with the compounds classification.
The goal is to send the petition to the FDA prior to the initiation
of Phase II.
As subsequent formulation development
proceeds, dissolution studies are done on new formulations in
accordance with the FDA guidance (3) and petitions are submitted
to the FDA requesting waivers of in vivo bioequivalence studies.
Use of the guidance has allowed the waiver of 4 human bioequivalence
trials and a savings of approximately 1 million dollars.
Beyond Biowaivers
While savings resulting from application of the BCS to obtain
waivers of in vivo bioequivalence studies are notable, they may
not represent the most significant benefit from application of
the BCS. Early high throughput screening information regarding
solubility and permeability should be invaluable to discovery
chemists. Previous authors have noted that compounds that have
good solubility and permeability are much more likely to lead
to an approved NDA then other compounds (9,10). Further, it has
been reported that 39% of compounds in development fail because
of poor biopharmaceutical properties (11). Thus consideration
of BCS criteria when selecting drug candidates may have a profound
impact by decreasing attrition during development.
References
1. Amidon, G.L., H. Lennernäs, V.P. Shah, and J. R.
Crison, "A theoretical basis for a biopharmaceutics Drug
Classification: The correlation of in vitro drug product dissolution
and in vivo bioavailability." Pharmaceutical Research 12
(3, March), 413-420, 1995.
2. Kaus, L.C., W.R. Gillespie, A.S. Hussain, and G.L. Amidon,
"The effect of in vivo dissolution, gastric emptying rate,
and intestinal transit time on the peak concentration and area-under-the-curve
of drugs with different gastrointestinal permeabilities."
Pharmaceutical Research 16 (2, February), 272-280, 1999.
3. United States Food and Drug Administration. Guidance
for Industry: Waiver of In Vivo Bioavailability and Bioequivalence
Studies for Immediate-Release Solid Oral Dosage Forms Based on
a Biopharmaceutics Classification System. August 2000. http://www.fda.gov/cder/guidance/3618fnl.htm
4. European Agency for the Evaluation of Medicinal Products.
Note for guidance on the investigation of bioavailability and
bioequivalence, (CPMP/EWP/QWP/1401/98), Committee for proprietary
medicinal products, July 26 2001. http://www.emea.eu.int/pdfs/human/ewp/140198en.pdf
5. United States Food and Drug Administration. CDER New
and Generic Drug Approvals: 1998-2002. http://www.fda.gov/cder/approval/index.htm
6. DiMasi JA. New drug development in the United States
from 1963 to 1999. Clin Pharmacol Ther 2001;69:286-96
7. United States Food and Drug Administration, Division
of Product Quality Research. Biopharmaceutics Research. January
16, 1998. http://www.fda.gov/cder/dpqr/dpqr_pgm.htm
8. Surendran N, Stilgenbauer LA, Michael S, Kibbey C, Woodhams
P, Stankovic C, Gifford E, Mandagere A, Quimby A, Wu S, Stewart
BH. Implementation of a Process for Early ADME/PK In Vitro Profiling
of New Chemical Entities in Support of Drug Discovery. AAPS PharmSci
2000 AAPS Annual Meeting Supplement. 2000; 2 (4) Available from:
http://www.aapspharmsci.org/scientificjournals/pharmsci/am_abstracts/2000/162.html.
9. Venkatesh S, Lipper RA. Role of the development scientist
in compound lead selection and optimization. J Pharm Sci. 2000
Feb; 89(2):145-54.
10. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental
and computational approaches to estimate solubility and permeability
in drug discovery and development settings. Adv Drug Deliv Rev.
2001 Mar 1; 46(1-3):3-26. (PII of original article: S0169-409X(96)00423-1.
The article was originally published in Advanced Drug Delivery
Reviews 1997 23:3-25).
11. Prentis RA, Lis Y, Walker SR. Pharmaceutical innovation by
the seven U.K.-owned pharmaceutical companies (1964-1985). Br
J Clin Pharmacol. 1988 Mar; 25(3):387-96.