 back
to text
back
to textInstitute of Pharmaceutical Technology,
Johann Wolfgang Goethe University, Frankfurt am Main, Germany
*email correspondence to: Sandra.Klein@em.uni-frankfurt.de
INTRODUCTION
Since the inclusion of the reciprocating cylinder dissolution
apparatus in the United States Pharmacopoeia (USP apparatus 3)
as an alternative to the basket and paddle apparatus for drug
release testing, very few studies with this set-up have been reported
in the literature. In particular there have been next to no studies
of the influence of changing the test parameters on drug release
rates of modified release (MR) formulations [1].
In principle, the USP apparatus 3 is most attractive for the
study of MR formulations, since changing the media to simulate
passage through the GI tract can be easily and reproducibly achieved.
For mesalazine (5-ASA) dosage forms used in the treatment of
Crohn´s disease and ulcerative colitis, it is important
to achieve high concentrations at the local inflamed areas while
minimizing systemic absorption. Release of drug proximal in the
GI tract (stomach and upper small intestine) should be avoided
to circumvent premature absorption and consequent drug wastage
and systemic side effects [2]. The current study was undertaken
to compare the in vitro dissolution characteristics of currently
marketed mesalazine dosage forms. The USP dissolution apparatus
3 was used to compare the dissolution behavior of four mesalazine
products available on the German market under conditions simulating
their transit through the GI tract in the fasted state.
Inflammatory Bowel Disease (IBD)
Inflammatory Bowel Disease (IBD) is a chronic and debilitating
illness prevalent in the Western population. It is characterized
by chronic intestinal inflammation that often shows an intermittent
course with acute attacks followed by periods of remission. Clinical
symptoms during acute attacks include diarrhea, bleeding, abdominal
pain, fever, joint pain, and weight loss. These symptoms can range
from mild to severe, and may gradually and subtly develop from
an initial minor discomfort, or may present themselves suddenly
in full-blown form. IBD can manifest itself in a variety of forms,
the most common of which are Crohn's disease and ulcerative colitis.
Both of these diseases can present very similarly in terms of
clinical symptoms, even though their inflammation patterns are
distributed differently in the GI tract.
Crohn's disease (CD)
Crohn´s disease is a chronic transmural inflammation of
the bowel which can affect the whole gastrointestinal tract, usually
in a discontinuous pattern. The initial location of CD is most
commonly in the lower ileum. From here the inflammation typically
spreads towards proximal parts of the small intestine. However,
the colon is also often involved. Depending on location and extension
of the disease, the clinical manifestations can vary markedly.
During the history of disease, numerous complications can occur,
with the result that approximately 70-80% of the patients will
undergo resective surgery in part of the GI tract during their
lifetime.
Currently it is not possible to cure Crohn´s disease.
The main objective of therapy is to contain the inflammation.
Therefore, treatment is dominated by anti-inflammatory drugs,
including corticosteroids and mesalazine (5-ASA), which are both
prescribed in all phases of the disease.
Ulcerative colitis (UC)
Ulcerative colitis is a chronic inflammatory bowel disease affecting
only the colon and shows a continuous distribution in the gastrointestinal
mucosa. In most patients the focal point of the inflammation is
in the distal part of the colon and the rectum. From this origin,
the inflammation often spreads proximally. In the most severe
cases, the whole colon is affected and one speaks of a "pancolitis".
About 30% of patients suffer from this severe form of UC.
As in the case of Crohn´s disease, it is not possible
to cure ulcerative colitis. General aims of treatment are therefore
to bring acute attacks into remission and thereafter to prevent
relapse. Here, too, medical treatment is dominated by anti-inflammatory
drugs.
Formulation of mesalazine dosage forms
Mesalazine plays an important role in the treatment of both Crohn´s
disease and ulcerative colitis. Its main principle of action is
a topical effect at the inflamed mucosa. Systemic absorption should
be minimized, as this leads to unwanted systemic side-effects
and inefficient redistribution of the mesalazine to the sites
of inflammation. Therefore, oral mesalazine dosage forms should
release the active substance selectively at the inflamed areas
in the gastrointestinal tract. Because of the different disease
patterns of Crohn´s disease and ulcerative colitis, different
formulations are required to adequately treat different patient
subgroups. Currently marketed formulation concepts include a)
tablets coated with gastric resistant pH-sensitive polymers and
b) microspheres which release the active drug via diffusion controlled
mechanism.
The aim of the present study was to evaluate the ability of the different formulations to release drug at various locations within the GI tract and subsequently to identify which formulations are suitable for which patient subgroups.
EXPERIMENTAL METHODS
Materials
Dissolution behavior was tested in different mesalazine oral dosage
forms, all of which were kindly donated by their manufacturers.
Three of them are approved in Germany for the treatment of both
CD and UC. The fourth dosage form (Asacolitin®) is currently
only approved for the treatment of UC. The test series included
the following products: Claversal® 250mg tablets (Ch-B.: 161726),
Merckle GmbH, Ulm; Salofalk® 250mg tablets (Ch-B.: 98H04918/E),
Dr. Falk Pharma GmbH, Freiburg; Pentasa® 500mg tablets (Ch-B.:
EI260T), Ferring Arzneimittel GmbH, Kiel; Asacolitin® 400mg
tablets (Ch-B.: 13323), Henning GmbH & Co., Berlin. Mesalazine
drug substance (lot# U60554) was purchased from Chemie S.p.A.,
Italy. All other chemicals were analytical grade or equivalent,
and purchased commercially.
As the formulations of the four products differ (see table 1), different release patterns are to be expected.
Table 1:
Marketed dosage forms used in the study and their formulation
concepts [10]
| Product | Dosage form | Polymer type* | Trademark of the polymer | Release pH |
| Claversal® | coated tablets | MA:MM (1:1) | Eudragit® L | > pH 6 |
| Salofalk® | coated tablets | MA:MM (1:1) | Eudragit® L | > pH 6 |
| Asacolitin® | coated tablets | MA:MM (1:2) | Eudragit® S | > pH 7 |
| Pentasa® | Coated microgranules | Ethylcellulose | Surelease® | pH independent release |
*MA= methacrylic acid, MM: methacrylate
Dissolution media and residence times
To simulate pH conditions along the GI tract, five different compendial
media were used (see table 2):
To characterize drug release at the primary site of systemic drug
absorption, the mid jejunum, a series of tests was performed in
SIFsp USP 24, pH 6.8. To compare drug release under different
pH conditions in the upper GI tract, further experiments were
performed in:
· SGFsp USP 24, pH 1.2, to simulate the fasted stomach
· Phosphate buffer, pH 7.2, to simulate proximal parts
of the ileum
· SIFsp USP 23, pH 7.5, to simulate the distal ileum.
To simulate the passage through stomach and the small intestine,
all dosage forms were additionally tested with a pH gradient method
based on mean physiological pH values in each segment [3]. In
the current test series a gastric residence time of 2 hours was
used. Although this value is rather long compared to the expected
gastric residence time in the fasted state for most of the dosage
forms studied, we wanted to evaluate the ability of three of the
products (Claversal®, Salofalk® and Asacolitin®) to
meet quality control criteria with respect to enteric coating
properties. For the simulation of fasted state residence times
in the different regions of the small intestine, averages of mean
transit times reported in several gamma-scintigraphy studies were
used [4,5,6].
Table 2: Dissolution media and
transit times used in the study
| GI Segment |
|
|
|
| Stomach |
|
|
|
| Duodenum |
|
|
|
| Jejunum |
|
|
|
| Proximal Ileum |
|
|
|
| Distal Ileum |
|
|
|
Dissolution test and sampling conditions
All drug release experiments were performed with a reciprocating
cylinder apparatus (BIO-DIS® RRT 8, CALEVA Ltd, Dorset, England).
The vessels were filled with 220 ml of media. The mesh size of
the top and bottom screens was fixed at 420 µm, which is
an intermediate value. A standard dip rate of 10 dpm was used
in all experiments [1].
Samples (3ml) were periodically withdrawn using a plastic syringe
(Henke-Sass, Wolf GmbH, Tuttlingen, Germany). The samples were
immediately filtered through a 0.45 µm Teflon filter (FP
030/2, Schleicher & Schuell GmbH, Dassel, Germany) and the
drug concentration was measured with a UV-spectrophotometer (U
2000, Hitachi Ltd, Tokyo, Japan). All studies were performed in
triplicate.
RESULTS AND DISCUSSION
In SGFsp (USP 24) no drug release was observed from the Eudragit®
L/S coated tablets (Claversal® 250mg, Salofalk® 250mg
and Asacolitin®). In contrast, about 60% of the drug was released
within 120 min under fasted state gastric conditions from the
Pentasa® microgranules. This excessive release in the stomach
would result in a relatively high loss of mesalazine to the systemic
circulation due to rapid absorption from the duodenum and subsequently
to less drug reaching inflamed tissue lower in the small intestine
and in the colon.
Testing in SIFsp (USP 24), a medium that reflects pH conditions
in the mid-jejunum, also leads to visible differences in drug
release behavior. In the case of Asacolitin®, the Eudragit®
S coated tablet indicated for the treatment of UC, no drug was
released within the experimental time-frame of five hours. On
the other hand, Claversal® 250mg and Salofalk® 250mg,
two tablet formulations with an Eudragit® L coating, both
released the active substance abruptly and quantitatively after
lag times of 30 and 150 min, respectively (see
figure 1).
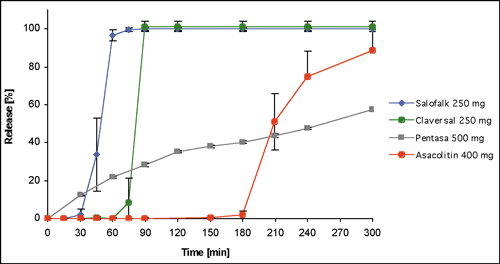
At first observation it seems remarkable that the lag times to onset of drug release differ so much from each other (about 120 min), because according to product descriptions Claversal® and Salofalk® have the same composition. In a series of tests Rudolph et al. [7] were nonetheless able to account for the observed differences. They noted that the onset of drug release correlates to erosion and subsequent leakage at the tablet edge. They then compared the film thickness of the two products at various locations on the tablets. In both Claversal® and Salofalk® the thinnest film coating was found at the tablet edge (approximately 100µm for Salofalk® and 250µm for Claversal®). The difference in the lag times correlated well with the difference in film thickness of the two formulations. Since for enteric coated products the characteristics of the coating are crucial for the onset of drug release, it is not unreasonable that the dissolution profiles strongly vary with the film thickness.
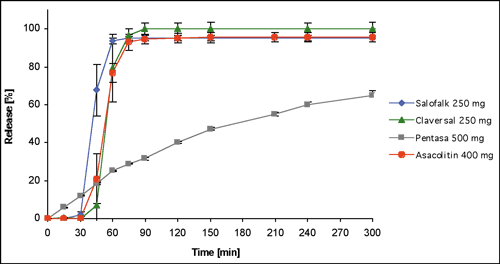
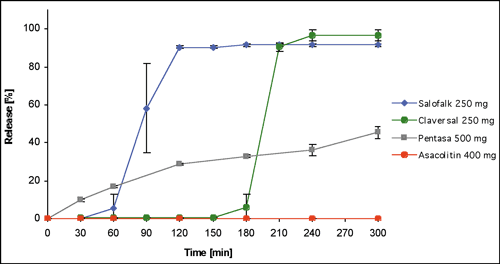
Increasing the pH of the test medium to pH values that are typical of conditions in the proximal and distal ileum leads to a convergence of dissolution profiles. At a pH of 7.2 the onset of drug release of Claversal® is delayed about 30 minutes compared with Salofalk® (see figure 2), but at pH 7.5 it is no longer possible to detect any difference between the two. Under these conditions even Asacolitin® (ostensibly formulated to release in the colon) shows the same dissolution behavior as Claversal® and Salofalk® (see figure 3).
Figure
1. : Dissolution behavior of different mesalazine dosage forms
in SIFsp pH 6.8 USP 24, expressed as mean + SD
back
to text
Figure 2: Dissolution behavior of different mesalazine dosage forms in Phosphate Buffer pH 7.2 Ph. Eur., expressed as mean + SD
Figure 3: Dissolution behavior of different mesalazine dosage forms in SIFsp pH 7.5 USP 23, expressed as mean + SD
Figures
1 to 3 indicate further that
the drug release behavior of Pentasa®, in contrast to release
from the enteric coated products, is influenced little by pH changes
within the small intestine. This is to be expected from its formulation,
which is based on a diffusion controlled release mechanism, and
the lack of pH dependence of mesalazine solubility in the intestinal
pH range.
The results described thus far, especially the results in SIFsp
pH 6.8, are useful illustrations of the clear cut differences
in drug release mechanisms. However it was obvious from the results
that it would be difficult to select a single set of test conditions
to adequately differentiate between the dosage forms.
As already mentioned, in the case of Crohn´s disease and ulcerative colitis it is important to achieve high concentrations of mesalazine at the inflamed sites of the mucosa without wastage of drug to the systemic circulation prior to reaching the inflamed area. Therefore it is reasonable to study release from the dosage form according to a method which can simulate its passage through the whole upper gastrointestinal tract. The use of a physiological based pH gradient in the Type 3 apparatus not only facilitates simulation of the upper GI transit within one experiment, but may also lead to more pertinent in vitro results since carryover effects can be detected. (A theoretical example of a carryover effect would be polymer swelling in gastric juice, which might lead to faster diffusion/erosion in subsequent media). Figure 4, shows the dissolution behavior of the four mesalazine dosage forms using the pH gradient method [3].
Figure 4: Dissolution behavior
of different mesalazine dosage forms (expressed as mean + SD)
during GI passage simulated using a pH gradient method [3]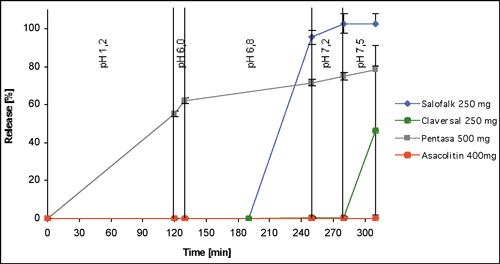
Results from the pH gradient method confirm that the three
pH sensitive acrylate-based formulations do not release under
gastric conditions within two hours. Since release from Pentasa®
is diffusion controlled, the Pentasa® microspheres release
a considerable amount of drug even in this medium. The higher
release rate at pH 1.2 than at intestinal pH values reflects the
higher solubility of mesalazine in acid solutions [7]. Assuming
a mean gastric emptying time of 60 min, as much as 40% of the
drug would be released under gastric conditions in the fasted
state. This release would lead to substantial drug absorption
in the proximal intestine and an increased risk of systemic side
effects. Furthermore, there will be a decrease in drug concentration
at the primary therapeutic target, namely the inflamed areas in
distal parts of the intestine and in the colon. For this reason
it seems that Pentasa® would be most useful in patients where
the inflammatory processes have spread to the more proximal parts
of the small intestine.
As expected, Asacolitin® shows no significant release under
simulated upper GI conditions and therefore appears to be more
suitable for the therapy of ulcerative colitis. To more thoroughly
evaluate its effectiveness in UC patients, it would be necessary
to extend the dissolution test procedure to include media that
reflect pH conditions in the colon, so that timely release of
the active drug at the inflamed sites in the colon can be verified.
In the case of Claversal® and Salofalk® tablets, both
coated with Eudragit® L, differences seen with tests in individual
media could be confirmed with a single test run using serial media.
Using the in vitro gradient method, mesalazine was abruptly released
from Salofalk® at pH values typical of the mid jejunum (pH
6.8). This may result in premature, high systemic absorption and
its associated disadvantages. Claversal® also exhibits an
abrupt release behavior but is distinguished from Salofalk by
the much longer lag time. On the basis of its in vitro behavior,
the Claversal tablet would be expected to reach the distal ileum
before releasing the active substance (see figure 4). This should
result in high concentrations of drug substance at the most inflamed
areas in case of both Crohn´s disease, where the inflammation
typically starts at the ileocecal valve, and in ulcerative colitis.
SUMMARY
Results from this study indicate that a USP apparatus 3 pH gradient
method is a convenient and discriminating method for comparing
the drug release behavior of controlled released dosage forms
during their passage through the GI tract. This dissolution method
is not confined to differentiating between formulations but could
also be used to examine the effects of inter- and intra-individual
variations in the gastrointestinal composition and transit of
both multiple and single unit dosage forms. Therefore the method
described should not only be applicable to compare marketed products
but also for the development of new MR delivery systems.
REFERENCES
1. Rohrs BR et al. USP dissolution apparatus 3 (reciprocating
cylinder): instrument parameter effects on drug release from sustained
release formulations. J Pharm Sci, 1995 Aug;84(8):922-6
2. Bondesen S., Intestinal fate of 5-aminosalicylic acid: regional
and systemic kinetic. Studies in relation to inflammatory bowel
disease preface, Pharmacol. Toxicol. 81 (Suppl. 2) (1997)
5-28[1]
3. Klein S. et al. Comparing drug release characteristics of different
marketed Mesalazine dosage forms by simulating their gi-passage
with a new BIO-DIS method (Abstract), Proceedings 28th International
Symposium on Controlled Release and Bioactive Materials, #6089,
Controlled Release Society, Inc., San Diego, California, USA,
2001
4. Davis SS et al. A comparative study of the gastrointestinal
transit of a pellet and tablet formulation. Int J Pharm, 1984,
21:167-177
5. Davis SS et al. Transit of pharmaceutical dosage forms through
the small intestine, Gut 1986 Aug;27(8):886-92
6. Coupe AJ et al. Variation in gastrointestinal transit of pharmaceutical
dosage forms in healthy subjects, Pharm Res 1991 Mar;8(3):360-4
7. Rudolph MW et al. A new 5-aminosalicylic acid multi-unit dosage
form for the therapy of ulcerative colitis. Eur J Pharm Biopharm.
2001: 183-190