
William Brown and Margareth Marques
The following questions
have been submitted by readers of Dissolution Technologies. Margareth
Marques, Ph. D. and Will Brown, United States Phamacopeia, authored
responses to each of the questions.
*Note: These are opinions and interpretations of the authors,
and are not necessarily the official viewpoints of the USP
Email for correspondence:
web@usp.org
Q
We have the following situation: a product
releases about 90% of label claim in 7 min in pH 6.8
phosphate buffer, about 40% of label claim in 60
min in 0.1 N hydrochloric acid, and about 50% of
label claim in 7 min and about 90% in 60 min in
pH 4.6 acetate buffer. Which of these media is the
most appropriate for this formulation?
A
The discrimination power of the dissolution test needs
to be considered when selecting the most appropriate
dissolution medium. Fast dissolution is not discriminative
enough and is appropriate only for drug substances
that are classified as BCS I (highly soluble and highly
permeable) when formulated in immediate-release
dosage forms. Another aspect to be considered is that the
dissolution test should be run in a feasible amount of time.
Based on the results obtained with 0.1 N HCl, it looks like it
will take some time to release 70�80% of the label claim. In
this situation, other parameters such as medium volume
and agitation speed could be modified to obtain a more
appropriate dissolution profile. The results obtained with
pH 4.6 acetate buffer look promising, but it is necessary to
verify which medium, hydrochloric acid or acetate buffer, is
more discriminative for the critical parameters of this
formulation and manufacturing process.
Q
Regarding the performance verification of USP
Apparatus 1 and 2 using the USP Salicylic Acid
Tablets RS, what is the reason to further dilute the
sample solution? Why can�t the tablet be dropped
and a direct reading obtained?
A
The procedure to verify the performance of USP
Apparatus 1 and 2 using the USP Salicylic Acid RS is
available at:
http://www.usp.org/referenceStandards/useAndStorage/calibrators.html
The procedure states,
"Measure the amount of salicylic acid dissolved from
filtered portions of the sample aliquots withdrawn and
suitably diluted with Dissolution Medium." This means
that it may be necessary to measure the absorbance of
samples that have been diluted so that the absorbance is
within the recommended range of 0.2�0.8 absorbance
units. In any method, for the diluted sample to be compat-
ible with the quantitative procedure being used, it must
be within the linear range of the analytical technique used.
If the absorbance of the undiluted sample aliquot is
acceptable, then no dilution is needed.
Q
When evaluating dissolution of capsules, must
the rotation speed be 100 rpm or is it possible to
use other speeds such as 50 or 75 rpm for baskets?
A
The agitation speed of the basket is selected by taking
into account how the particles are being dispersed in the
dissolution medium and the discrimination power of the
test regarding the critical parameters for the formulation
under evaluation. It is not advisable to use speeds above
100 rpm unless justified to give results that reflect in vivo
performance or to provide better discrimination without
adversely affecting method reproducibility. The decision
to use a particular rotation speed (or other test conditions)
will be based on the evaluation of dissolution profiles
obtained under various conditions.
Q
Please describe the calculation of the amount
released when using an open-loop USP Apparatus 4
(flow-through cell).
A
If an online UV spectrophotometer is being used, the
sample absorbance is measured at regular intervals, the
concentration calculated, and the dissolution rate
determined as the multiplication product of the
concentration and the flow rate. This release rate at each
sampling time can be reported, or the cumulative
amounts dissolved can be estimated for each sampling
interval. The cumulative amount dissolved can be
estimated from the average dissolution rate at the
beginning and ending time points multiplied by the
elapsed time (trapezoid rule). The calculations can be done
by hand or by using a spreadsheet.
Alternatively, the cumulative amount dissolved can be
measured exactly using an off-line spectrophotometer.
The drug concentration in the fraction collected over the
entire interval is the most accurate measure of the amount
of drug released. Furthermore, the volume collected gives
a better confirmation of the actual cell flow rate during
the interval.
Q
Could the USP Salicylic Acid Tablets RS be used to
qualify the USP Apparatus 4 (flow-through cell)?
A
Up to now, the USP Salicylic Acid Tablets RS have not
been fully validated to verify the performance of USP
Apparatus 4. There are papers in the literature, such as
Kauffman, J. S. Qualification and Validation of USP
Apparatus 4. Dissolution Technol. 2005, 12 (3), 41�43, that
have evaluated the use of this USP Reference Standard in
the qualification and performance evaluation of this
apparatus, but this use it is not official or compendial.
Q
What could be the volume of dissolution medium
for the evaluation of drug release from buccal
patches or tablets?
A
The minimum volume of dissolution medium to be
used in the evaluation of drug release from any dosage
form is defined by taking into account the sink condition
of the drug substance. As the volume of fluid in the buccal
pouch is considerably less than the typical volumes
encountered in dissolution testing of tablets and capsules,
a system that can provide dissolution results based on
similar volumes might be useful in the evaluation of the
performance of buccal patches or tablets during dosage
form development. Several designs for small-volume
dissolution test apparatus are available. The most
important concern for any dissolution test is that the set
conditions are sensitive to product changes that may
adversely affect the therapeutic effect.
Q
Is there a minimum amount of drug to be
released when defining the Q value for a particular
dosage form?
A
No, there is no minimum amount to be released. During
the development of an immediate-release product, a
target Q of about 70�80% released in 30�45 minutes is
commonly used until more information about the product
becomes available. The final definition of the Q value is
going to be made after considering the dissolution
profiles of all batches (pilot, manufacturing, stability) and
the probability of passing the S1, S2, S3 criteria defined in
the USP General Chapter <711> Dissolution. More
information can be found the in following papers: (1)
Dumont, M. L., Berry, M. R., Nickerson, B. Probability of
passing dissolution acceptance criteria for an immediate
release tablet. J. Pharm. Biom. Anal. 2007, 44, 79�84; (2)
Hauck, W., et al. Oral dosage form performance tests: new
dissolution approaches. Pharm. Res. 2005, 22 (2), 182�187;
and (3) Cholayudth, P. Using the Bergum method and MS
Excel to determine the probability of passing USP
dissolution test. Pharm. Technol. 2006, 30 (1), 88�94.
Q
What is the difference between artificial gastric
fluid and simulated gastric fluid?
A
Various formulas can be found for dissolution media
that approximate gastric fluid. Where a reference is made
to USP, the preparation of simulated gastric fluid can be
found under Test solutions, in the USP section Reagents,
Indicators, and Solutions.
Q
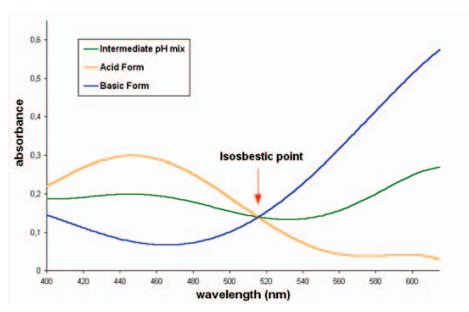
What are the reasons for quantifying the amount
of drug release at the isosbestic point and not at
the wavelength of maximum absorption?
A
Isosbestic point is a specific wavelength at which
two or more chemical species have the same
concentration-based absorptivity. When superimposing
the absorption spectra of these two species at equivalent
concentrations (molar or mg/mL), the isosbestic point
corresponds to the wavelength at which these spectra
cross each other. An example is shown above in the
spectrum of bromocresol green (source: Wikipedia).
Isosbestic points are useful in quantifying the amount of
drug released when the dissolved drug undergoes
degradation into a defined product, such as aspirin, which
hydrolyzes to salicylic acid (see Aspirin Tablets monograph
in USP 31).
Q
Is it acceptable for a tablet or capsule to float
during the dissolution test? Must the floating
dosage form be weighed down with a sinker to
keep it at the bottom of the vessel? Must the
dosage form sink to the bottom of the vessel?
Must a sinker be used?
A
The dosage form should lie at the bottom of the vessel
during dissolution testing with the paddle apparatus. The
various regions in the vessel are associated with different
characteristic hydrodynamics, and consistency of results is
more likely when the sample remains in place. A floating
sample may move into areas of high or low flow within the
medium and may encounter the moving paddle, leading
to a mechanical interaction that may impair the integrity
of the dosage form. Any of these events may alter the
dissolution rate of the individual unit under test and
increase the variability of the results. The use of a sinker is
often included in a method description and occasionally
part of the monograph procedure. The sinker design has a
great influence in the dissolution profile of the dosage
form. A number of different types of sinkers are available,
and when sinkers are used, the particular design
employed should be well described in the method
documentation.
Q
I am testing an extended-release product using
the criteria given in the dissolution general chapter.
The procedure calls for sampling at three time
points, 1, 4, and 8 h. For the first six tablets tested,
the results met the criteria in Acceptance Table 2 at
the 1- and 8-hour time points but did not meet the
criteria at the 4-hour time. When we repeated the
test with the next six tablets, the results were
acceptable at each time point. Did the product pass
the test at L1 for some time points and at L2 for the
remaining time point?
A
he performance of the product is measured over all
time points. Under the Interpretation section of the
general chapter, the instruction is to continue testing
through the three levels unless results conform at either
L1 or L2. In the case given, the results did not conform at
L1 (six units tested) and you continued to L2 testing (six
additional units for a total of twelve). The procedure calls
for sampling at each time point, which you did. Having
results for all time points, you compared with the
acceptance table. All results for L2 testing are used if
testing needs to proceed to that level.