
William Brown and Margareth Marques
The following questions
have been submitted by readers of Dissolution Technologies. Margareth
Marques, Ph. D. and Will Brown, United States Phamacopeia, authored
responses to each of the questions.
*Note: These are opinions and interpretations of the authors,
and are not necessarily the official viewpoints of the USP
Email for correspondence:
web@usp.org
Q
USP says that for multiple sampling times, the sample aliquot should be replaced with an equal volume of fresh medium, but if the replacement is not necessary, the calculations should correct for the volume change. Is there a calculation to correct the results when we replace the sample volume? What is the correction if we do not replace the sample volume?
A
A dissolution test with multiple sampling times will produce erroneous, inaccurate results if the amount of sample (medium volume or dissolved drug substance) removed at early sampling times is not considered in calculations of results for subsequent sampling times. If a significant amount of dissolved drug is removed, it should then be included in the calculation of total amount dissolved at each subsequent time. The decision of significance is a matter of interpretation, but the decision should be documented and should become part of the standard procedure. The effect of the removal of medium volume is an additional consideration. In the calculation of dissolved drug, the medium volume is a scaling
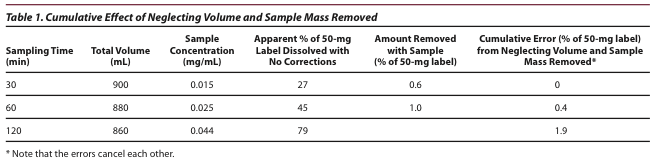
factor from concentration to total amount in solution. If the sampling volume taken is not replaced, then the actual remaining volume must be used in calculations of total amount dissolved. Table 1 illustrates the cumulative effect of neglecting the volume removed (no replacement) and dissolved drug removed over three sampling times with 20 mL removed at each time point. As you can see in this example, the error from the amount of drug removed somewhat cancels the error in assuming no loss of volume. In summary, accurate dissolution results will represent the total mass dissolved, whether present in the medium or taken for analysis, and use the actual volume of medium at the time of sampling.
Q
We are planning to produce a nifedipine extended- release tablet. The USP monograph has eight tests
for dissolution, each with different dissolution conditions. We found that some of the formulations failed to pass one or more of the tests while others succeeded, and we are confused. Which test should we depend on? Must the formulation pass all of the tests, or is one test enough?
A
We may find tempting the notion that because products may have similar doses and dosing intervals, they should have the same dissolution test. In the present state of the art, that is simply not the case for extended-release products. The USP Nifedipine Extended-Release Tablets monograph represents at least two products, Adalat CC and Procardia XL, with distinctly different controlled- release mechanisms. Similarly, the United States market has four generic products for Adalat CC and three generic products for Procardia XL (see FDA Orange Book at http://www.accessdata.fda.gov/scripts/cder/ob/docs/queryai.cfm). While bioequivalence is used to establish generic status, the release mechanisms of reference listed drugs and generics may not produce adequately similar profiles in the dissolution test conditions to satisfy one quality control performance test. All of that being said, a product for which a regulatory judgment on bioequivalence has not been given may not perform acceptably in any of the published test methods such that the dissolution criterion, percent dissolved in a set time, may not be satisfied. Depending on your use of the dissolution test, you may need to develop a test specifically for the product in hand. Currently, the best measure of bioequivalence is through a true human study, and except for very narrowly defined conditions, in vitro testing alone may not be the best indicator.
Q
Some of our results from the USP PVT with prednisone tablets were outside the upper limit for combined geometric mean. Otherwise, the com- bined geometric mean and the percent CV met the
requirements. How is it possible to have individual results out of range but still pass the test?
A
The standard for the PVT uses summary statistics as the basis for the criteria. Thus, the geometric mean and not the individual results is used for comparison against the ranges given. As you might agree, the mean will summa- rize a range of values, some of which may be outside the criteria. However, the standard is specific for the mean, and if the underlying sample has some values out of the range, that is of no consequence. The other component of the new PVT criteria is the percent CV. This new control on the width of the sample range will encourage the use of dissolution test assemblies that produce similar results (internally). In effect, it now makes the assembly behave like a unit.
Q
In Figure 2 of the USP General Chapter <711> Dissolution, the dimension for the base of the paddle is 35.8 ± 1.0 mm. In the Japanese Pharmacopoeia (JP), it is 35.6 ± 1.0 mm for the same part of the apparatus. Is there an explanation for this discrepancy since this general chapter is harmonized among USP, the JP, and the European Pharmacopoeia (EP)?
A
A discrepancy existed between the USP and the global harmonized dissolution chapter figure found in JP XV for the paddle and the radius. The value that was agreed to by the signatories (USP, JP, and EP) was 35.8 ± 1.0 mm. The JP text has been amended in Supplement 1 of JP XV.
Q
I am working on a new product (tablet) dissolu- tion method development using USP Apparatus 2 (paddle). In this regard, I need one clarification; may I use any rpm other than 50, 75, and 100 rpm? For example, if I use 60 rpm, is it a deviation from the regulatory requirements?
A
You can use any rotation speed that is appropriate to demonstrate the discriminatory power of the dissolution test. Rates outside the range of 25-150 rpm are usually inappropriate because of the inconsistency of hydrody- namics below 25 rpm and because of turbulence above 150 rpm.
Q
How many USP monographs have more than one dissolution or drug release test?
A
There are two ways this information can be obtained. The most laborious one is a manual search in the printed version of USP-NF. The easiest way is a search of the USP- NF electronic version. However, because we had to use unusual keywords, we cannot be sure that the number we found is accurate. We thought that the most accurate search would use the string "more than one dissolution" because monographs with more than one dissolution test have the following statement in the Labeling section:
"When more than one Dissolution test is given, the labeling states the Dissolution test used only if Test 1 is not used." Using this strategy, we found that 54 monographs have more than one dissolution test. It is easier for drug release tests, because with the harmonization of the General Chapter Dissolution in USP, EP and JP, drug release tests now only apply to transdermal systems. There are two monographs with more than one drug release test.
Q
Which edition of USP first introduced the concept of dissolution specifications for delayed- and extended-release products?
A
In the Pharmacopeial Forum (PF) 1985, 11 (4), six proposed monographs for extended-release products were published. See the Headquarters column for that issue of PF. A history of the policy on modified-release standards appears in the PF 1980-1985 on pages 2262, 2383-2384, 2991, and 2999. The Preamble/Preface to USP XXI (1985) discusses the inclusion of modified-release performance standards. USP XXI also includes General Chapter <724> Drug Release for the first time. The first monographs for extended-release products are found in the Second Supplement to USP XXI: Potassium Chloride Extended-Release Capsules and Potassium Chloride Extended-Release Tablets, official on July 1, 1985. These were followed in the Fifth Supplement to USP XXI (official on May 15, 1987) with at least six new monographs for extended- and delayed-release products.
Q
The USP Performance Verification Test (PVT) calculation tool (available at http://www.usp.org/ USPNF/pvtToolStatement.html) states that the valid use of PVT requires that the choice of single- or two-stage testing be made before beginning testing. Does this mean we should automatically run 12 vessels to be sure? We initially run six tablets, and when we use the calculation tool, the test results indicate that Stage 2 must be done. Will the initial data be invalidated since Stage 2 was conducted after the first set of data was examined? The terms in the Calculation tool are quite confus- ing. Stage 1 calls for 12 results, and Stage 2 requires 6 results.
A
The certificate that is provided with the USP Predni- sone Tablets RS (available at http://www.usp.org/ referenceStandards/useAndStorage/calibrators.html) gives the instructions for the procedure and the calcula- tion of the results for the PVT. USP provides an online tool that simplifies the calculation of the results. It is not necessary to use the online tool, and the results can be readily calculated by hand. The PVT can be performed as either a single-stage procedure or a two-stage procedure. For 6-, 7-, and 8-position test assemblies, the single-stage procedure requires two runs (e.g., 12, 14, or 16 results, respectively). When the results have been obtained from
the two runs, the geometric mean and percent CV are calculated and compared with the allowed ranges. The two-stage procedure allows the laboratory to stop the testing after the analysis of the first run. In the two-stage procedure, if the resulting geometric mean and percent CV do not conform with the allowed ranges after the first
run, an additional run is performed (second stage). The results from both runs are used to determine the combined geometric mean and percent CV to compare with the allowed ranges. The choice of doing the single- stage procedure is up to the laboratory. The choice must be made prior to the collection of results.