Questions and Answers February 2020
Margareth R. Marques, Ph.D. and Mark Liddell, Ph.D.
The following questions have been submitted by readers of Dissolution Technologies. Margareth R. Marques, Ph.D., and Mark Liddell, Ph.D., United
States Pharmacopeia (USP), authored responses to each of the questions. *Note: These are opinions and interpretations of the authors and are not
necessarily the official viewpoints of the USP.
Email for correspondence:
mrm@usp.org
Q Can the validation of accuracy in a dissolution method be carried out in volumetric flasks or in the dissolution equipment?
A The validation of any analytical procedure, including dissolution, should be carried out in conditions that closely represent those of the actual procedure. Therefore, the validation of any dissolution method should be performed using the dissolution equipment that will be used for the actual procedure.
Q How is the validation of precision and accuracy performed for a dissolution test that, according to the USP monograph, is carried out using pooled sampling?
A Pooled sampling is typically used for routine analysis such as batch release. For most other types of evaluation, including validation, the test should be done with unit sampling, and the validation follows the recommendations in the USP general chapter <1092> The Dissolution Procedure: Development and Validation.
Q Is it necessary to calibrate USP Apparatus 1 when a 10-mesh basket is being used?
A The term “calibration” is not useful in the context of the performance qualification of a dissolution test assembly. USP uses the term “performance verification test” (PVT), when referring to a demonstration of acceptable performance for a dissolution test assembly. The collaborative study used to establish the acceptance ranges for the USP PVT material employs 40-mesh baskets. It is simply not practical for USP to provide dissolution ranges for the PVT for all conceivable variations on the standard dissolution apparatus. A typical approach is to use the standard 40-mesh baskets to assure that the other components of the test assembly are functioning properly by performing the USP PVT. Provided that the 10-mesh baskets are in good physical condition, one might be able to assert that the test assembly should then also be suitable for use with 10-mesh baskets; however, USP does not provide specific acceptance criteria to determine apparatus suitability for baskets other than the standard 40-mesh baskets used in the PVT collaborative study.
Q Is the shaft height check in USP Apparatus 1 and 2 done only during mechanical verification, or is it done only for the performance verification test?
A The mechanical verification should be performed prior to the execution of the performance verification test. In practice, it is also recommended that the height of the shaft in USP Apparatus 1 and 2 should be checked every time the dissolution equipment is assembled. Depending on the type of shafts installed in the instrument and their likelihood to come out of adjustment, checking the shaft height regularly may be necessary.
Q Is there any recommendation for when disks should be used in the disintegration test?
A The use of the disks in the disintegration test is decided on a case-by-case basis, supported by data obtained from the samples being evaluated.
Q We are using the US FDA dissolution methods database to review products that are considered extended-release formulations to assist with selecting dissolution conditions. However, the monographs, for the most part, do not appear in the USP-NF. Can they be found in another location?
A The FDA dissolution methods database is for products for which there is no USP monograph available. There is also a USP dissolution database that complements the FDA database, where you can see the dissolution, disintegration, and drug release conditions stated in the USP monographs. The USP database is available at https://www.usp.org/resources/dissolution-methods-database
Q How is the height of 160-210 mm measured for 1L vessels, as stated in the USP general chapter <711> Dissolution?
A The USP general chapter <711> does not specify whether the height is specific to the overall height of the vessel or the height of the inner volume of the vessel. Given that the chapter states “for a nominal capacity of 1L” and specifies the range of acceptable inside diameters, there is a wide range of acceptable vessel heights. USP has reported that when the vessel height is measured from the bottom of the flange to the inside bottom of the vessel, as shown below, some vessels currently available on the market do not meet the USP specifications (Liddell, et al. Dissolution Technol. 2007, 14, 28-33. DOI: 10.14227/DT140107P28). However, as previously explained, this specification may not be critical to the operation of the instrument as long as the shaft height is set using the inside bottom of the vessel as the reference point. 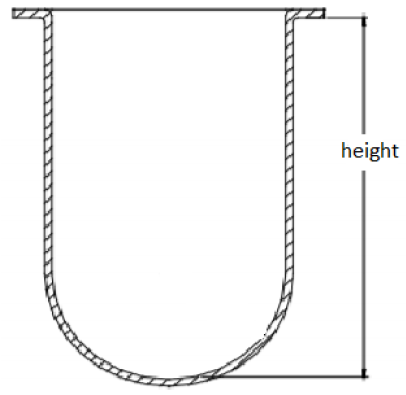
Q Is it necessary to perform forced degradation during the validation of selectivity for a dissolution test?
A Forced degradation is done to evaluate the specificity of analytical methods when there is no reference material for the impurities or degradation products. In the case of dissolution testing, forced degradation is not necessary, but you need to evaluate if there is any degradation of drug substance during the dissolution test and how this degradation is going to be taken into consideration. One example is the case of aspirin, which degrades during the dissolution test and measurements are carried out or done at the isosbestic point where both aspirin and salicylic acid are present. Another example is the case of tacrolimus, where there is formation of its epimer, and the amount of tacrolimus and epimer are both taken into account when calculating the amount of tacrolimus released in the dissolution test.
Q Is it preferable to use purified water or ultrapure water in the dissolution test?
A Always start with purified water. Any other water quality grade needs to be evaluated in a case-by-case approach, and it may be formulation dependent.
Q When conducting the dissolution test 2 as stated in the USP monograph for Estradiol Transdermal Systems, which uses phosphate buffer with sodium lauryl sulfate as dissolution medium, the presence of a white precipitate was observed. What could be a possible reason for the presence of this precipitate in the dissolution medium?
A One possibility may be the due to the incompatibility of sodium lauryl sulfate (SLS) with potassium salts. Buffers containing SLS should be prepared using the sodium salt of the reagent.
Q We are developing a dissolution test for immediaterelease tablets with a functional score, and we would like to know how to evaluate the results. Should the three stages be used from the Acceptance Table 1 in USP general chapter <711> Dissolution?
A The dissolution results for tablets with functional score are evaluated according to the instructions in the USP general chapter <705> Quality Attributes of Tablets Labeled as Having a Functional Score, where 12 split tablet portions are evaluated. The average of the 12 results is not less than Q, and no single result is less than Q - 15%. Both Q and the 15% are calculated based on the product label claim.
Q What could be the acceptance criteria for the disintegration test of capsules when there is no compendial monograph?
A The USP general chapter <701> Disintegration does not provide a default test time for most dosage forms. There is awareness that capsule shell opening is an important step in bioavailability. Unintended delay in capsule shell opening could be a cause for concern. Typically, the dissolution test is a more comprehensive evaluation of drug availability in vitro, incorporating information beyond capsule shell opening time. The recommendation is to first develop a dissolution test and then, depending on the physical-chemical characteristics of the drug substance and the dissolution profile of the drug product, the dissolution test may be replaced with a disintegration test, with appropriate justification. The acceptance criteria for disintegration are developed on a case-by-case approach considering results from the samples being evaluated. See the proposal for the new USP general chapter <1711> Oral Dosage Forms - Performance Tests published in Pharmacopeial Forum 45(6), available at www.usppf.com.
Q If an extended-release product does not meet the L1 stage acceptance criteria as stated in the USP general chapter <711> Dissolution, should it be considered a deviation and treated accordingly?
A No. If the product fails L1, the testing should proceedto stages L2 and L3. A product is considered to not meet the dissolution acceptance criteria if it fails stage L3. Then, the appropriate procedures for an out-of-specification (OOS) investigation should be conducted.
Q The acceptance criteria for the L1 stage for extended-release dosage forms states “no individual value lies outside each of the stated ranges.” For six capsules tested, the individual value should be read as each capsule value or as the mean of six sample values?
A The requirements are met if the quantities of active ingredient dissolved from each of the individual dosage units tested conforms to the acceptance criteria. Therefore, at each time point there will be six results, one from each individual unit tested that must conform with the requirements of the test.
Q In the USP general chapter <711> Dissolution, there are instructions on how to add pepsin and pancreatin to the dissolution medium, but the amount of these enzymes differs from the specifications for using simulated gastric fluid TS and for simulated intestinal fluid TS (TS = test solution). How do we select which amounts should be used in the dissolution medium?
A The amount of enzyme to be used depends on the purpose of the test. The amounts stated in the USP general chapter <711> Dissolution are used when there is evidence of cross-linking observed in dissolution tests with gelatin capsules or gelatin-coated tablets. Simulated gastric fluid with or without pepsin and simulated intestinal fluid are options for dissolution tests where a biorelevant medium may be required. These reagents, simulated gastric and intestinal fluids, should not be used when investigating cases where gelatin cross-linking is a suspected cause of a gelatin-containing dosage form failing to meet dissolution criteria.