
STIMULI TO THE REVISION PROCESS
Stimuli articles do not necessarily reflect the policies of the USPC or the USP Council of Experts
Revision of the Dissolution Procedure: Development and Validation <1092>
Subcommittee on The Dissolution Procedure: Development and Validation <1092> to the
Pharmaceutical Dosage Forms Expert Committee:
R Skwierczynski1, P Curry, V Gray, J Krämer, E Stippler, J Suggett, T Mirza2, and W Brown3
1Ray Skwierczynski, Chair.
2Tahseen Mirza, FDA Liaison.
3Correspondence should be addressed to Will Brown, Senior Scientific Liaison, US Pharmacopeial Convention, 12601 Twinbrook Parkway, Rockville, MD 20852; tel. 301.816.8380.
ABSTRACT
In this Stimuli article a Subcommittee of the Pharmaceutical Dosage Forms Expert Committee discusses a proposed revision to general information chapter The Dissolution Procedure: Development and Validation <1092>. Published elsewhere in this issue of PF, the proposed revision provides a new structure that divides the process of development and validation of the dissolution test into its component parts. The revision adds sections about preliminary assessments needed before initiating method development and about automation. A new section, Interpretation, within Acceptance Criteria clarifies the interpretation of results from the dissolution test. The relationship between bioavailability, bioequivalence, and the dissolution test is considered in general chapter Assessment of Drug Product Performance—Bioavailability, Bioequivalence, and Dissolution <1090>.
BACKGROUND
The dissolution test is an important means of assuring the continuing performance of non-solution orally administered drug products. The development of a dissolution test procedure is briefly discussed in general information chapter In Vitro and In Vivo Evaluation of Dosage Forms <1088>, whereas general information chapter Validation of Compendial Procedures <1225> gives limited validation information for dissolution testing. Neither of these two chapters provides a level of detail and focus sufficient for dissolution testing. In 2001, a Stimuli article provided an initial rationale and discussion of content for a new general information chapter (1). The new chapter, The Dissolution Procedure: Development and Validation <1092>, was intended to supplement the information in <1088> and <1225> and provided step-by-step detail for both development and validation as well as offering information on new technology and equipment. In 2006, the chapter became official with the Second Supplement to USP 29-NF 24 (2-4).
The General Chapters—Dosage Forms Expert Committee 2010-2015 placed the review and possible revision of The Dissolution Procedure: Development and Validation <1092> on its work plan for the 2010-2015 revision cycle (2011) (5). A subcommittee was formed in 2011 to carry out this task, which is reflected in the proposed revision appearing elsewhere in this issue of PF.
SCOPE AND PURPOSE
Non-solution orally administered dosage forms are the preponderant dosage forms that need in vitro performance testing. General chapter Oral Drug Products— Product Quality Tests <2> recommends inclusion of the dissolution test as the primary performance test for drug products. The Subcommittee deliberated on the scope of the revision of The Dissolution Procedure: Development and Validation <1092> and determined that performance testing of solid oral dosage forms should continue to be the main emphasis. Many of the observations and suggestions made have applicability to products delivered through other routes of administration such as topical, injectable, and mucosal dosage forms.
The title of the chapter suggests that the dissolution test comprises a single procedure that can be developed and validated. However, the dissolution test actually requires two procedures to be performed sequentially. The sample preparation procedure is usually seen as the dissolution test. That procedure is followed by quantification of the dissolved drug in the presence of excipients and dissolution medium, which in this chapter is termed analytical finish. The development and validation of a dissolution test involves consideration of each of the subordinate procedures separately and also their inter-relationship. On review, the Subcommittee concluded that the current chapter did not clearly differentiate between issues relating to the dissolution portion of the test from those that arise from the development and validation of the analytical procedure.
ORGANIZATION OF PROPOSED REVISION
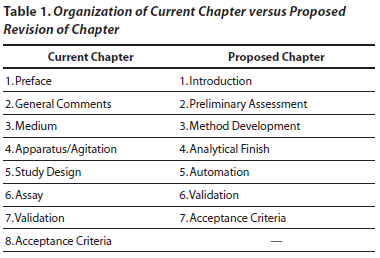
The proposed revision of the chapter is divided into sections that follow the time sequence of activities involved in the preliminary assessment, and development of the procedures for the dissolution method and for the analytical finish. The proposed revised chapter then addresses the validation of the combined procedures. By contrast, the current chapter does not clearly lay out an order of events but rather concentrates on each aspect, regardless of the stage. In addition, the proposed revision of The Dissolution Procedure: Development and Validation <1092> includes new sections on automation and on the acceptance criteria and their interpretation. Table 1 shows the sections in the current chapter and proposed revision, to allow for comparison.
PRELIMINARY ASSESSMENT
Although the Preliminary Assessment section is new in the proposed revision of the chapter, it draws on existing text. This new section discusses the activities that need to be performed before a dissolution test is developed. A basic understanding of the physicochemical properties of the drug substance is a necessary precondition for development of the dissolution test. To that end, the subsections address determination of the drug substance solubility and stability in aqueous media, and also information on filtration and filter selection. Information is provided on the reasons for choosing the apparatus, medium, and volume.
Determining the solubility and stability of drug substances in various media at 37 °C is an important component of the preliminary assessment. The proposed revision includes suggested procedures and test considerations. An important enhancement to the content of the proposed revision is a listing of commonly used surfactants with values, when available, for the associated critical micelle concentrations. Cautionary statements about the quality of the surfactant used as well as any known interactions of the surfactant with components of common buffers are included. New to the chapter is specific advice on the conditions of interest in stability studies. As in the current chapter, the final assessment of stability is part of the validation of the method.
Filters are discussed in greater detail than in the current chapter. An expanded discussion of issues that arise from adsorption onto the filter material, leachables from the filter, and filter pretreatment is provided. Special topics are introduced, such as the use of filters in Apparatus 4 testing and the use of filters in automated sampling.
Choosing an apparatus, medium, and volume are the final activities leading to development of the dissolution procedure. These sections discuss the considerations necessary for making these choices. The proposed revision includes new information about the need for a surfactant in the medium, as well as its concentration. It also includes a more in-depth discussion of the use of media that represent more closely the composition of stomach and intestinal fluids in the context of developing an in vitro-in vivo correlation (IVIVC). This discussion now occurs under the section on choice of media. The possible problem of acid stage testing of delayed-release dosage forms, where the drug is poorly soluble or unstable in the acid stage, is mentioned as a consideration. A listing of apparatus used in the performance testing of semi-solid dosage forms has been added.
METHOD DEVELOPMENT
The term “method development” here refers to the procedures resulting in the sample to be analyzed and represents the in vitro performance of the dosage form. Proposed additions to the chapter include:
- For the section on deaeration, addition of an example of recent information on the measurement of the level of deaeration related to a particular product.
- For the section on sinkers, addition of cautionary statements on their use and also a mention that sinkers can have uses other than buoyancy correction.
- For the section on agitation, conditions for Apparatuses 3 and 4 are provided. The notion that laminar or turbulent conditions can be obtained for Apparatus 4 is discussed, based on new information derived from the literature.
- For the section on data handling, addition of a methodological discussion of results calculation and discussions about cumulative versus fractional dissolution rates and handling the data from pooled dissolution.
The section, Study Design, is subdivided into discussions of time points, observations, and sampling as in the current chapter. The subsection on filters is moved to the Preliminary Assessment section and centrifugation is discussed under Analytical Finish in the proposed revision. The discussion of sampling time points in the proposed revision includes additional information on the use of the f2 similarity factor for profile comparison. Information on the timing issues associated with delayed-release product testing is also provided. The list of common observations in the current chapter is increased in the proposed revision, with additional examples. The current chapter includes autosampling under the general topic, Sampling. In the proposed revision of the chapter, autosampling has been placed under the new, overarching topic of Automation. The discussion of issues relating to manual sampling has been broadened to include positioning and medium replacement. A new section, Dissolution Method Assessment, closes the method development section with advice on the suitability of the dissolution method.
ANALYTICAL FINISH
The analysis of the dissolution samples begins with the sampling process. These analyses are most commonly conducted using ultraviolet and visible spectrophotometry or high-performance liquid chromatography (HPLC) procedures.
AUTOMATION
Automation is a new section on a topic that was addressed only briefly in the current chapter (autosampling). Different modes, such as on-line and off-line analysis, and examples of different instrument configurations are presented. This section discusses both advantages and deficiencies of automation. New sections related to automation, such as medium preparation, sample introduction and timing, sampling and filtration, cleaning, operating software, and computation of results are included. Common deviations from compendial procedures, which may require validation, are also offered as part of this section.
VALIDATION
This section builds on the information in the existing chapter. A reference to Validation of Compendial Procedures <1225> and ICH documents helps to update this section from the current chapter. It allows flexibility in the use of samples from the intact drug product, spiked placebo, or simulated dissolution samples, depending on the validation parameter of interest. The proposed revision provides a new definition of the blank, as the term is applied to dissolution testing. In the section on accuracy and recovery testing, reference is made to the filter assessment that is part of the preliminary activities. The proposed revision also mentions that reproducibility is generally an extension of intermediate precision involving different analysts at separate laboratories. Design of experiments methodologies are mentioned as useful tools in robustness testing, and reference is also made to Validation of Compendial Procedures <1225> with parameters associated with the robustness of the analytical finish. A proposed new chapter concerning statistical tools for validation is scheduled for PF 40(2) and may also be useful.
ACCEPTANCE CRITERIA
This section is greatly enhanced, compared with the information given in the current chapter. Sections on immediate-release, delayed-release, and extendedrelease dosage forms, explain the approaches to criteria for dissolution testing of these various products. Multiple dissolution tests are sometimes found within a single monograph, and therefore an explanation of this situation is offered. Reference is made to General Notices, section 4.10.10. Applicability of Test Procedures, which provides the general principles involved.
The interpretation of dissolution results, as explained in Dissolution <711> under the section Interpretation, is presented. The Acceptance Tables represent immediate release, the two stages of delayed-release testing, and extended release. The interpretation of dissolution results can be a confusing process and has been the subject of many queries directed to USP staff. The criteria are explained using hypothetical tolerances.
REFERENCES
- Gray, V. A.; Brown, C. K.; Dressman, J. B.; Leeson, L. J. Stimuli to the Revision Process. A new general chapter on dissolution. Pharm. Forum 2001, 27 (6), 3432-3439.
- <1092> The Dissolution Procedure: Development and Validation, Preview. Pharm. Forum 2004, 30 (1), 351-363.
- <1092> The Dissolution Procedure: Development and Validation <1092>, In-Process Revision. Pharm. Forum 2005, 31 (5), 1463.
- <1092> The Dissolution Procedure: Development and Validation. In The United States Pharmacopeia and National Formulary USP 29-NF 24, Second Supplement; The United States Pharmacopeial Convention, Inc.: Rockville, MD, 2006.
- de Mars, S.; Abernethy, D. R.; Koch, W. F.; Long, A. G.; DeStefano, A. J.; Williams, R. L. General chapter management in the 2010-2015 cycle. USP General Chapter Project Team, Prescription-Nonprescription Stakeholder Forum. Pharm. Forum 2009, 35 (5), 1372- 1379.