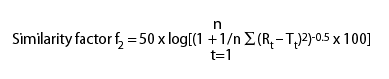
Meeting Report:
"AAPS
Workshop on SUPACs: Where are We
Going? Have We Reached the Limit?
David C. Pang
Director of
Scientific Affairs, American Association of Pharmceutical Scientists
AAPS conducted a workshop on Scale-Up
Post Approval Changes (SUPACs), March 15-16, 1999, Crystal Gateway
Marriott, Arlington, VA. Some of the goals of the workshop were:
1) to review the scientific, regulatory and quality basis of SUPACs,
2) to assess if SUPACs are current with advancement in science
and regulatory policy; and 3) to discuss successes and failures
of SUPACs and current issues.
The SUPAC initiatives were started
to provide scientific rationale to expedite the processes of post
approval changes of drug products so that FDA can assure their
safety and effectiveness and at the same time lower the regulatory
burden for industry. Problem areas were identified and research
was performed at academia, industry and FDA laboratories to support
changes in 1) components and composition; 2) manufacturing (process
and equipment); 3) scale of manufacture; and 4) site of manufacture.
The specifications for the drug product include sterility, dissolution
rate, containers and closure systems. It was the intent of the
research to improve specification requirements and modify testing
procedures for the product quality aspects of drug production.
The changes may shorten the application process for industry and
facilitate the approval process with the FDA.
Investigators at University of Maryland/Baltimore,
FDA and pharmaceutical industry gathered data on the impact of
scale-up, formulation, and other manufacturing changes on product
quality, resulting in the release of a number of guidances by
FDA, including: 1) Immediate release solid oral dosage forms,
scale-up and post-approval changes: chemistry, manufacturing and
controls, in vitro dissolution testing, and in vivo bioequivalence
documentation (SUPAC-IR); 2) Modified release solid oral dosage
forms, scale-up and post-approval changes: chemistry, manufacturing
and controls, in vitro dissolution testing, and in vivo bioequivalence
documentation (SUPAC-MR); 3) Semi-solid dosage forms, scale-up
and post approval changes: chemistry, manufacturing and controls,
in vitro dissolution testing, and in vivo bioequivalence documentation
(SUPAC-SS), 4) Intermediates in drug substances synthesis/bulk
actives post approval changes: chemistry, manufacturing, and controls
documentation (BACPAC I) and 5) SUPAC-IR/MR manufacturing equipment
addendum. Several other SUPACs are at various stages of development
including one for transdermal delivery systems (SUPAC-TDS) and
one for sterile aqueous solutions (PAC-SAS). As can be deduced,
the guidances have been developed on the basis of types of dosage
forms.
The guidances provide specific recommendations
on documentation of equivalence in post approval changes of drug
products in component-composition, batch size, manufacturing process
and site of manufacture. Each category is divided into 2 or 3
levels based on the degree and type of changes in the approved
product. Post-change product batch is considered the same as the
pre-change product batch when there is no discrepancies in their
dissolution profiles or the release rates. The similarity in the
physicochemical properties of the batches from the release test
or dissolution profile can be used to infer that the pre-change
products and post-change products are also the same in terms of
safety, efficacy or bioequivalence.
SUPAC-IR uses in vitro dissolution
tests to indicate product sameness. Dissolution profile comparison
on post-change batch and pre-change batch is indicated by the
similarity factor, f2.
In vitro-in vivo correlation is an
important measure for SUPAC-MR involving development, validation,
application and dissolution profile comparison. In vitro release
test is used in SUPAC-SS as a measure of product quality and sameness.
The equivalence approach based on a standard confidence interval
procedure is utilized to verify that the lots are close enough
for product sameness between pre-change and post-change batches.
The International Society for Pharmaceutical
Engineers (ISPE) collaborated with FDA to provide equipment equivalence
lists which clearly define classes and subclasses of equipment
by unit operation. The Equipment Addendum Guidance documents provide
assistance for changes in equipment.
Further updating of the SUPACs is
possible based on the approach delineated in FDAMA Section 116
to permit pre-approved supplements, change-being-effected supplements
and annual report filings. The concept of SUPAC on drug product
is being extended to drug substance (BACPAC I and II). The general
approach works to achieve continuing pharmaceutical equivalence
and bioequivalence after approval for NDAs and ANDAs, as well
as other approved FDA products.
There were 1,039 SUPAC supplements
submitted for generics and 263 for new drugs from 1995 to 1998.
Immediate release and alternate site changes dominated the topics
for SUPAC supplements with 582 and 431 submissions, respectively,
for generic drugs and 142 and 66, respectively, for new drugs.
Semi-solids and modified release dosage forms accounted for less
than 30 submissions for both types of drugs during the three year
period. Based on qualitative estimates provided by pharmaceutical
company representatives, consulting experts, and the number of
SUPAC-IR submissions during 1997, the Office of Planning and Evaluation,
FDA, estimated savings for 1997 chemistry, manufacturing and control
changes to be $70.7 million. The savings generated by SUPAC were
realized through shorter waiting times for site transfers, more
rapid implementation of process and equipment changes, more rapid
implementation of batch size increase, production of fewer unmarketable
stability test batches, and reduced stability tests and reduced
administrative cost. For example, there was documentation that
the times taken for one company in submissions and reviews of
supplements on packaging and manufacturing were 600 and 111 months,
respectively, before and after the adoption of SUPAC guidances.
Overall, several industrial representatives
at the workshop stated that there were no major issues with SUPAC.
The coordination at CDER was excellent. After the initial learning
period, pharmaceutical companies and the review divisions and
district offices of FDA were comfortable with the processes involved
in SUPAC.
It was the consensus of the scientists
at the SUPAC workshop that there are several issues that need
to be addressed for future SUPAC applications. The topics that
need to be addressed are dosage form independent issues, SUPAC
IR/MR/SS issues, site transfer, and equipment process changes.
Dosage form independent issues include multiple changes, packages,
and sizes. SUPAC-IR/MR issues include: 1) mechanism to control
changes to specification and in-process control; 2) capsules (size,
shapes, color); 3) tablet weight; 4) shape, size, scoring, energizing;
5) need to simplify Case C dissolution; 6) product-specific selection
of media; and 7) f2 specification/profile. Site transfer issues
include:1) harmonization of IR/MR site transfer requirements for
stability; 2) transfer of experiment date to new site, and 3)
the need for a bioequivalent test for level 3 SUPAC-MR. Equipment
process change issues include 1) the use of biopharmaceutical
classification system to justify changes being effected (CBE)
and 2) clarification of deposition of other types of level 2 and
3 changes. SUPAC-SS issues include preservatives and in vitro
release testing.
As stated by Larry Augsburger, President of AAPS, the range of dissolution profiles of products found bioequivalent in the Maryland study may be broad enough to encompass many major changes. Consideration should be given to: 1) reducing the dissolution requirement for Class I drugs. For the examples studied, the requirement of 85% dissolution in 15 minutes was not justified, 2) reducing the filing requirements for a change in technical grade of excipient at least to CBE classification in accordance with 21 CFR 314.70(c). Though limited in the number and classes of excipients tested, the Maryland data do not support Level 2 (PAC), 3) shifting of levels downward by an increment of one for compositional changes, 4) removing reference to 10X in re scale changes, and 5) reducing the media required in Level C dissolution tests to two or three.