Questions and Answers May 2025
Margareth R. Marques, Ph.D. and Mark Liddell, Ph.D.
The following questions have been submitted by readers of Dissolution Technologies. Margareth R. Marques, Ph.D., and Mark Liddell, Ph.D., United
States Pharmacopeia (USP), authored responses to each of the questions. *Note: These are opinions and interpretations of the authors and are not
necessarily the official viewpoints of the USP.
Email for correspondence:
mrm@usp.org
Q If a stability study for a gelatin capsule product fails to meet Tier I specification at a specified interval due to cross-linking, for instance at 9 months, should the remainder of the stability study (months 12-18) be conducted the Tier I testing at each timepoint or can a Tier II analysis be performed immediately?
A Once the process of cross-linking of the gelatin starts, it does not stop even when the agent causing the cross- linking is removed. Therefore, during stability studies, once cross-linking in the gelatin is discovered, all remaining stability study dissolution tests should be conducted with the addition of enzymes in the dissolution media. See USP general chapter <1094> Capsules - Dissolution Testing and Related Quality Attributes.
Q When should dissolution testing with and without the addition of enzymes (Tier 1 and Tier 2, respectively) be done? Are both tests required for routine analysis for product batch release?
A Gelatin, in the presence of certain compounds, mainly aldehydes, and in high temperature/high humidity storage conditions, can have cross-linking between the gelatin chains. When this occurs, the gelatin becomes insoluble in aqueous solvents. Enzymes can be added to the dissolution medium to aid in digesting the cross-linked gelatin allowing the capsule contents to be exposed to the dissolution medium. The enzyme is selected based on the pH of the dissolution medium. The enzymes can only be added to the dissolution medium if there is evidence of the presence of cross-linking in the gelatin. See USP general chapters <711> Dissolution and <1094> Capsules - Dissolution Testing and Related Quality Attributes for more information about cross-linking. The addition of enzymes to the dissolution medium can be done at any time, during routine batch release testing and/or stability studies, essentially any time there is evidence of cross- linking during dissolution testing.
Q Regarding the use of USP Apparatus 4 (flow-through cell), if a 1-mL of aliquot was withdrawn at each time point using an integrated automatic sampling system, should the volume removed be replaced? The equipment was connected to a 500-mL media reservoir, which is located on a heated stir plate. The stir plate was set at 37 °C. Is the hot-stirring plate enough to maintain the apparatus at 37 °C? Or should the hot plate be set at higher temperature to keep the apparatus at 37 °C?
A USP Apparatus 4 provides continuous flow of dissolution medium through a flow-through cell containing the dosage form or test sample. Consequently, there is no medium replacement because the medium is continuously passing through flow-through cell and across the sample surface during the entire duration of the test; however, the removed volume should be accounted for in the calculation of the cumulative amount released. Regarding the temperature control, it is up to the lab to provide data demonstrating that the use of the hot plate can maintain the temperature of the medium within the specified range, e.g., 37 ± 0.5 °C, during the entire duration of the test. Most apparatus 4 dissolution systems are equipped with a mechanism to preheat the media before it enters the flow-through cell.
Q Are chewable tablets exempt from the disintegration test?
A There is an FDA guidance recommending which tests are required for chewable tablets, which includes disintegration and dissolution tests. The link to FDA guidance can be found in USP general chapter <1711> Oral Dosage Forms - Performance Tests.
Q Which is the most appropriate manual sampling procedure in dissolution testing, the use of cannula or pipette?
A The recommendation is to use a cannula bent in an L shape (Fig. 1A) to facilitate the sampling in all vessels and most importantly to allow sampling from the appropriate sampling zone in the dissolution vessel. Generally, proper use of a pipette requires it to be held in a vertical orientation. Using a syringe and cannula allows flexibility in positioning of the cannula. Keep in mind that the agitation should not be stopped for the sampling. Once the sample has been withdrawn from the vessel, the cannula should be removed, and the sample should be filtered as soon as possible (Fig 1B). The purpose of filtration is to stop the dissolution process by separating undissolved particles from the dissolution media. When collecting multiple samples, each sample or set of samples must be filtered before collecting the next sample. 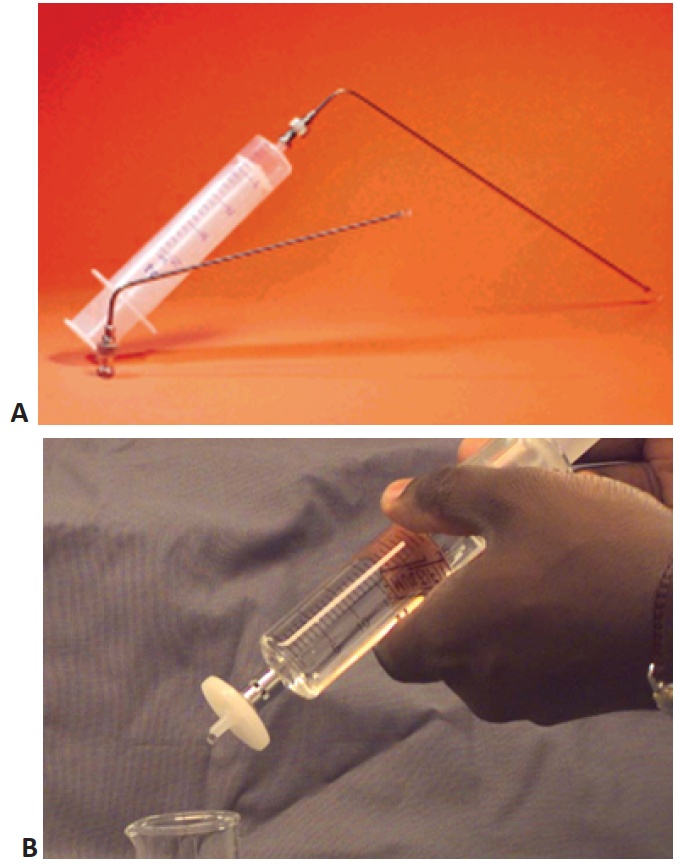
Q When should a laboratory investigation be triggered if there are out-of-specification (OOS) results in the dissolution testing; is it after the initial failure at L1 stage or after completing all three stages (L1, L2, L3)?
A The three stages stated in Acceptance Table 1 in USP general chapter <711> Dissolution are part of the dissolution test. An investigation of OOS results should be done if the batch under analysis fails L3. Ultimately, it is up to the individual manufacturers and internal quality policy to determine when an OOS investigation is initiated.
Q In USP general chapter <701> Disintegration, the acceptance criteria for uncoated tablets is provided as per individual monograph. If the monograph is not available for a particular product, what limit/acceptance criteria is recommended? If a dissolution testing is done for a particular product that was not official in USP, is it required to keep the disintegration test in product specifications?
A When developing a product, a dissolution test should be developed. The dissolution test may be replaced with a disintegration test depending on the physical-chemical characteristics of the drug substance and on the release mechanism of the dosage form. See USP general chapter <1711> Oral Dosage Forms - Performance Tests for more guidance. The disintegration acceptance criteria are defined on a case-by-case approach based on the results obtained with the samples under evaluation. The only products that require both dissolution and disintegration tests are chewable tablets and orally disintegrating tablets.
Q In USP general chapter <1092> The Dissolution Procedure: Development and Validation, specifically section 5.3 on Accuracy/Recovery, it states that “to enhance drug solubility it may be appropriate to prepare a stock solution by dissolving the drug substance in a small amount of organic solvent and diluting to the final concentration with dissolution medium.” Should this be performed exactly as described in the dissolution method?
A Any analytical method must be validated in conditions as close as possible to the conditions that are going to be used during the routine analysis. For example, if a solution is diluted as part of the dissolution method, the validation should be performed including the dilution step. This is applicable for any analytical method validation procedure. In the specific example cited above, the dissolution media solution is spiked with a concentrated solution of drug substance for the purpose of determining the accuracy/recovery of the analytical method when the concentration of the drug substance is measured in the presence of constituents that are present in the dissolution sample solution(s); therefore, the dissolution sample solution should be prepared as it is in the proposed dissolution method.
Q We are developing a dissolution test for a gelatin capsule containing a drug substance that is insoluble in aqueous solvents. Thus, a surfactant was added to the dissolution medium. We know that when there is evidence of cross-linking in the gelatin capsule, the appropriate enzyme must be added to the dissolution medium. USP general chapter <1094> Capsules - Dissolution Testing and Related Quality Attributes recommends a pre-treatment when the dissolution medium contains surfactant because it can denature the enzyme. The text specifies that the duration of this pre-treatment is not more than 15 min; however, this is not enough time to dissolve the drug substance. Should this time be extended?
A The pre-treatment stage allows the enzyme to initiate digestion of the cross-linked gelatin without the interference of other chemicals, i.e., surfactants, that may interfere with the enzyme activity. During this pre-treatment stage, the drug substance will not be in contact with the medium for the entire 15 min. The cross-linked gelatin needs to be digested by the enzyme to allow the capsule to rupture and the contents to be in contact with the dissolution medium. The 15-min pre-treatment time stated time in the USP chapter is a recommendation. Your laboratory should evaluate the most appropriate length of the pre-treatment stage for the product being evaluated. Also, check the activity of the enzyme being used. The activity must be determined by the procedure stated in the USP general chapter <711> Dissolution. The enzyme activity depends on the substrate being used.
Q We are using disintegration testing as an in-process control for tablet compression during manufacturing. Would you provide guidance regarding the typical disintegration time?
A The disintegration time for any type of product is determined in a case-by-case approach using data obtained with the samples being evaluated. The definition of this time should be discussed with your research/ development/formulation and manufacturing groups.
Q Are there any special recommendations regarding the use of enzymes in the dissolution medium when using the USP apparatus 3 (reciprocating cylinder)?
A There are no special recommendations for the use of enzymes in the dissolution medium using USP apparatus 3. The composition of the dissolution medium is determined by considering the composition and characteristics of the sample under evaluation and not the equipment.
Q What type of dissolution medium should be used with vegetable capsule shells?
A The composition of the dissolution medium is defined considering the physical and chemical characteristics of the dosage form and drug substance, mainly the solubility of the drug substance and capsule contents. Special attention should be given to capsules that are coated to provide a modified-release mechanism. In such cases, the composition of the coating must be considered.